发布者:抗性基因网 时间:2018-12-14 浏览量:1000
|
文 献 信 息 |
|
Ruppé E, Ghozlane A, Tap J, et al. Prediction of the intestinal resistome by a three-dimensional structure-based method[J]. Nature microbiology, 2018: 1. |
|
研 究 背 景 |
|
肠道微生物群被认为是抗生素抗性决定簇(ARD)的主要储库,ARD可能通过移动遗传元件转移至细菌病原体。然而,由于已知的ARD(主要来自可培养的细菌)与来自肠道微生物群的ARD之间的远距离同源性,这一假设得不到经验证据的支持。因此,尚未完全确定肠道ARD(即肠道抵抗组织)的准确普查。为此,在三维结构(同源比较模型)的基础上开发并验证了一种注释方法(称为成对比较模型),从而预测了来自人类肠道微生物群中390万种蛋白质中的6,095种ARD。
|
|
方 法 |
|
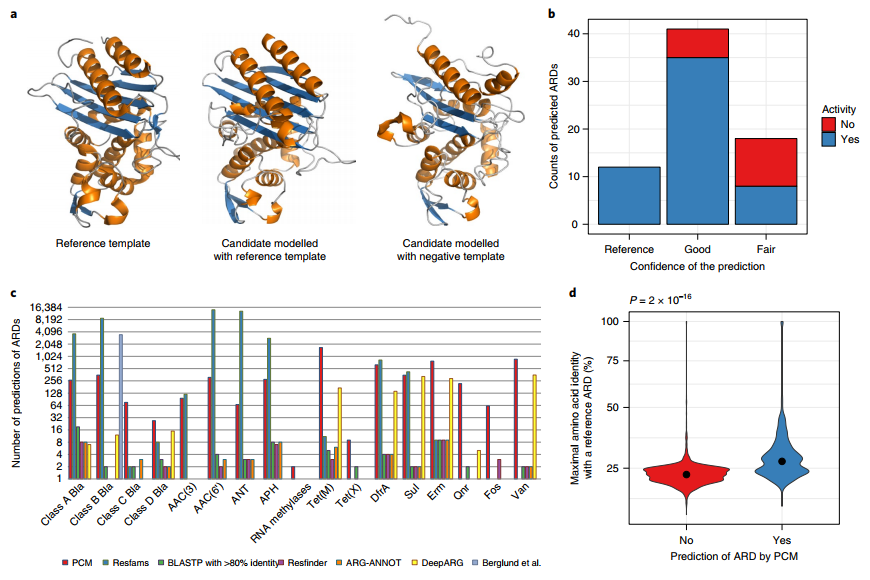
为了预测肠道微生物群中的ARD,我们开发了一种基于蛋白质同源建模的方法,我们称之为成对比较建模(PCM)。
|
|
研 究 结 果 |
|
我们开发了一种方法,PCM,可以揭示肠道微生物群中ARD的多样性。使用这个工具,我们收集的证据表明,我们预测的绝大多数急性呼吸道疾病没有表现出活动迹象,而且它们的丰度与基因丰富度相关。结合一些肠道细菌对抗生素的保护作用,我们的研究结果表明肠道微生物群中的急性呼吸道疾病可能被认为是我们的“抵抗力”,确保在抗生素暴露下保持健康的普通微生物群。
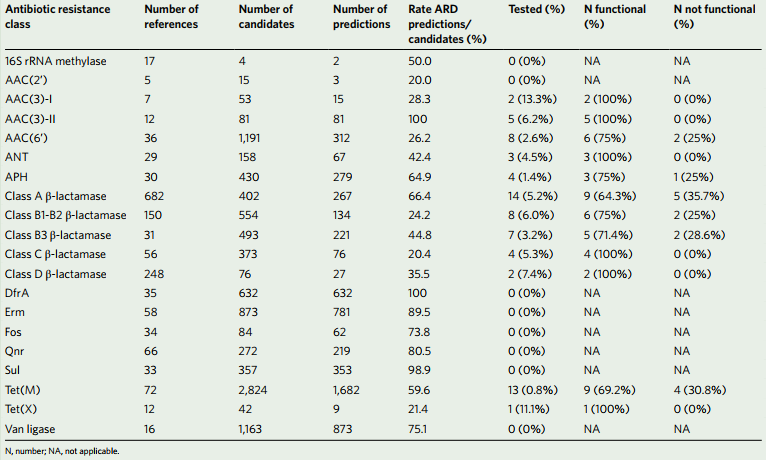
肠道微生物群中ARD的预测
表1.来自肠道微生物群19的390万基因目录和基因合成结果的ARD预测总结
|
|
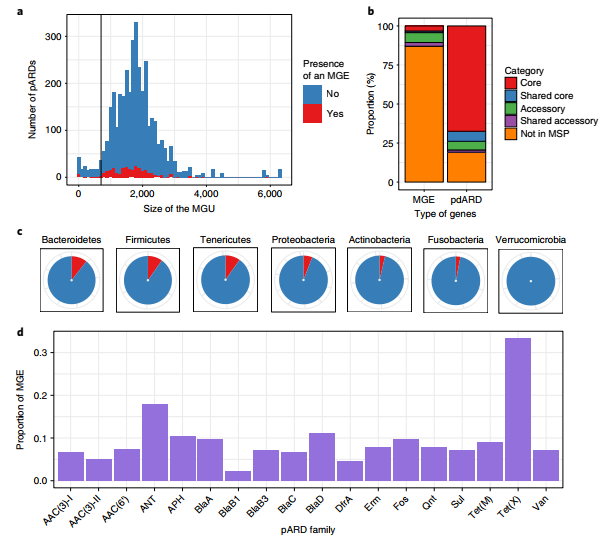
pdARD的定位以及与MGE的关联
|
|
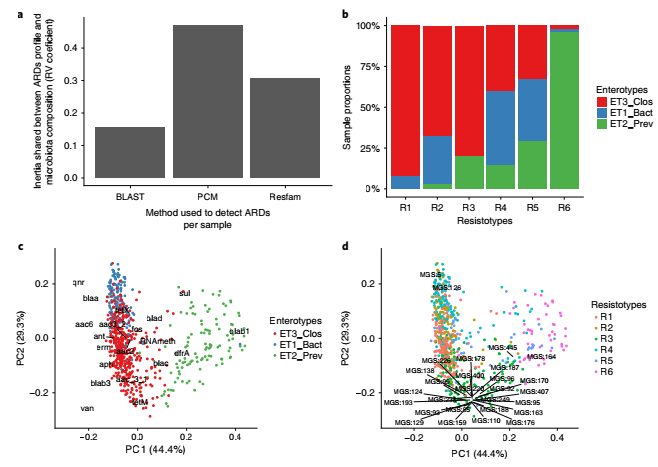
pdARDs在人体微生物群中的分布
图3. Metahit队列中663名个体的耐药类型、肠道类型、MGS和PDARds之间的关联。
|
|
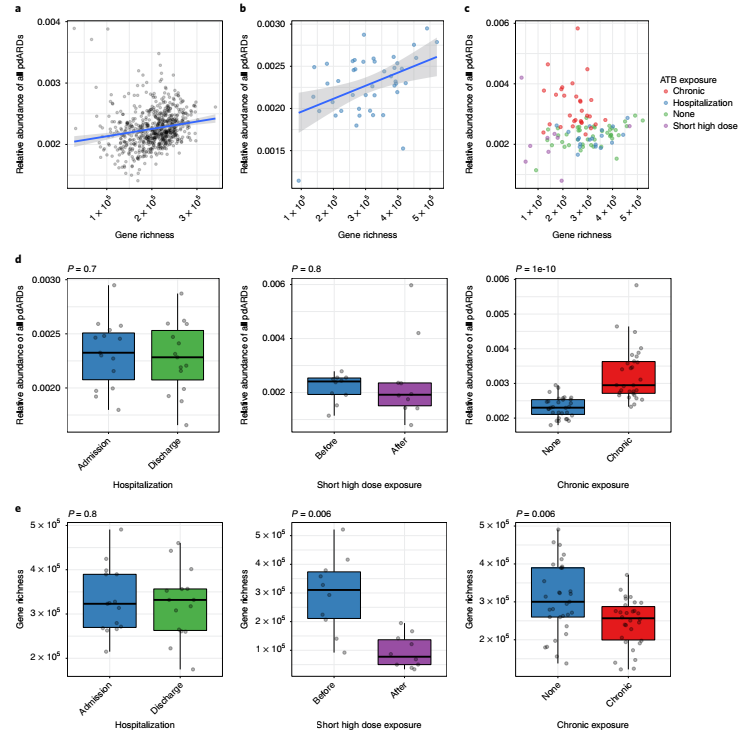
不同抗生素暴露下的pdARD动态
|
|
结 果 总 结: |
|
大多数肠道微生物群ARD可被认为是占优势的共生微生物群的固有特征,并且这些基因很少与细菌病原体共享。 |